P1: Evolvability, Epistasis and Molecular Robustness in Protein Evolution
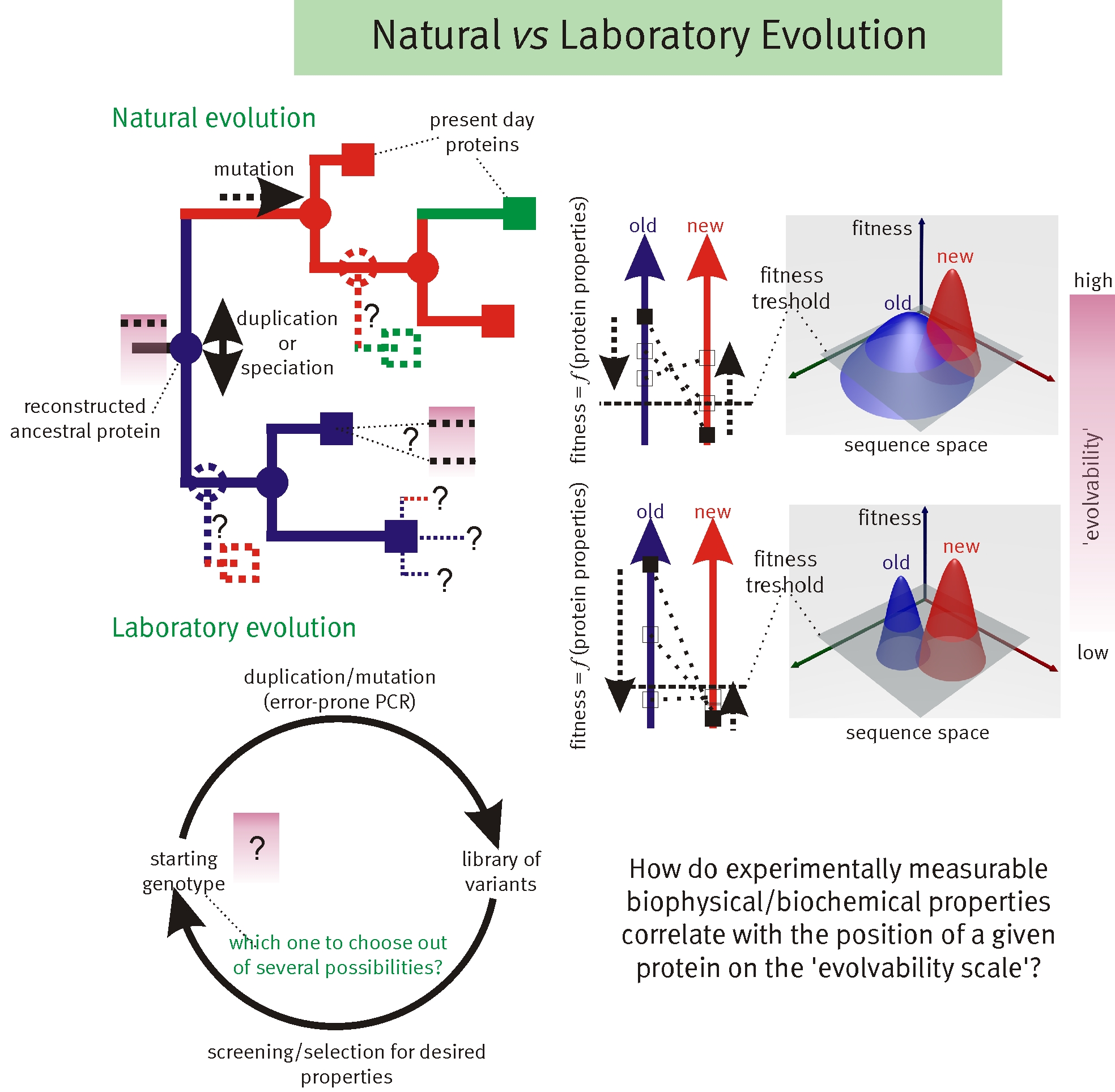
A long-standing riddle in molecular evolution concerns the question of how proteins, forming so many shapes and exercising so many fundamentally
different functions, can evolve into new proteins, with new structures and functions. Several models try to explain such innovations without depriving
an organism of its existing, possibly essential function exerted by the "old" protein. The most popular models include neofunctionalization,
subfunctionalization (SUBF) by degenerative mutations, and dosage models, all focusing on adaptation after gene duplication. “Escape-from-Adaptive-Conflict”
partially resolves this riddle by including adaptive processes before and after gene duplication that led to multifunctional proteins (promiscuous enzymes), and divergence (SUBF).
However, potentially beneficial changes are vastly outnumbered by those that are detrimental. Therefore, any protein also has to be robust to
mutation. The success of this balancing act between adaptability and the maintenance of general functionality ultimately determines which
mutations are fixed during adaptive evolution. In other words: how do evolving genes (or their encoded proteins) change their dominant and/or
latent function(s) without ever getting stuck in fitness minima?
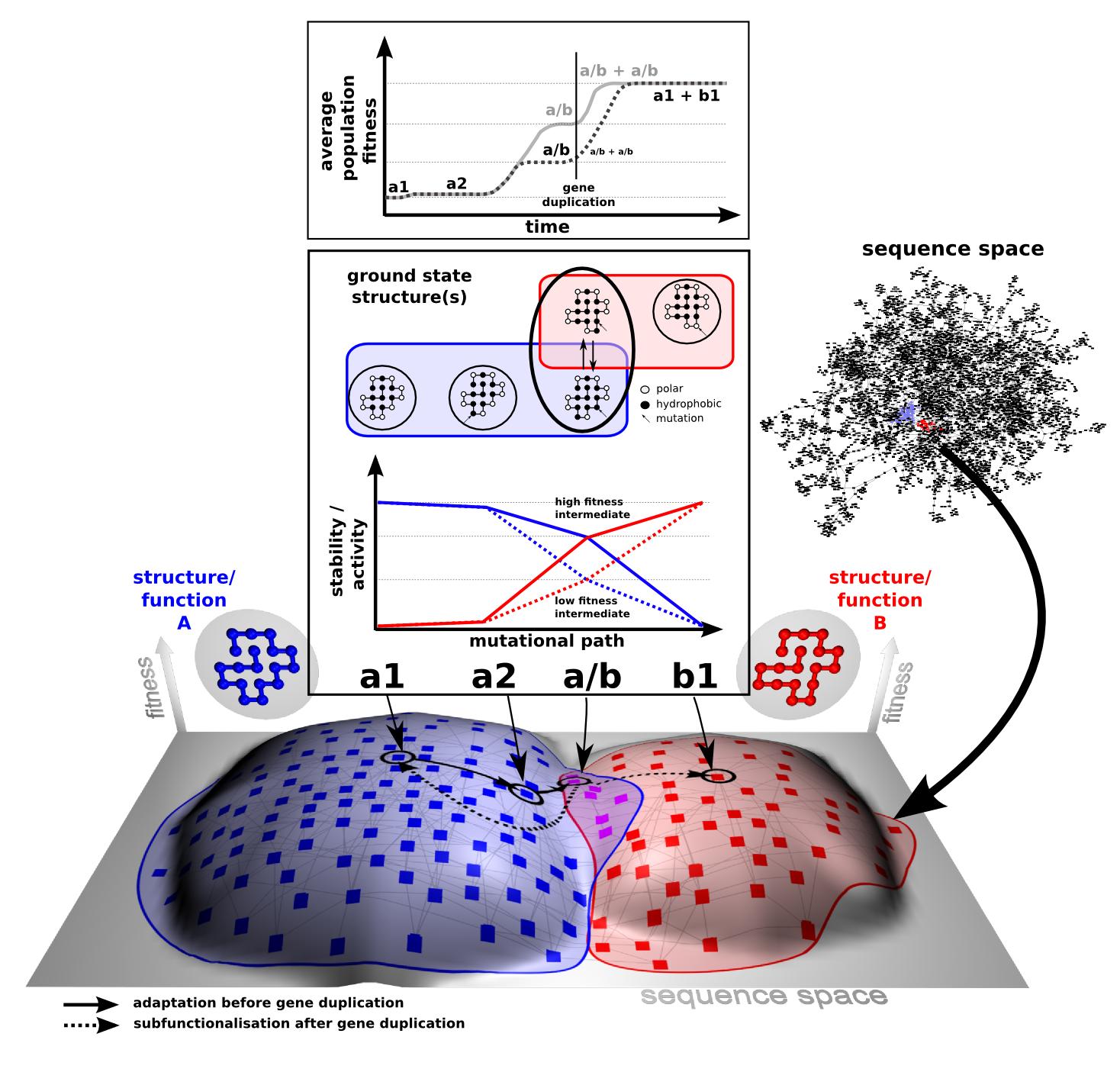
Recent results illustrate the importance of sub-optimal or promiscuous functions for the adaptation toward new function of protein coding genes. Unfortunately, this renders any modelling of fitness landscapes (and therefore rational design) incredibly complicated. We developed a theoretical framework that uses biophysical principles to infer the roles of functional promiscuity, gene dosage, gene duplication, point mutations, and selection pressures.
We find that selection pressures and duplication rates alone can determine which scenario will prevail. Multi-functionality becomes a crucial advantage when gene duplications are rare and an increase in mutational robustness, not necessarily functional optimization, can be the sole driving force behind SUBF. Overall, this is the first model in which all three processes are unified and demonstrates that, given a certain rate of gene duplications and point mutations, selection pressure determines which processes will prevail. Furthermore, by mapping both RNA and protein-like models on a unified landscape with tunable neighborhood properties, we demonstrate that the relationship between robustness and evolvability depends critically on the ratio of viable mutations which are neutral (coding the same phenotype) and innovative (coding for a new phenotype).
People: Berndjan Eenink, Margaux Aubel
Collaborations
- Hue Sun Chan (Toronto)
- Florian Hollfelder (Cambridge UK)
- Nobuhiko Tokuriki (Vancouver)
- Joachim Jose (Münster)
- Jürgen Pleiss (Stuttgart)
Funding: BBSRC (2002 - 2005), DAAD (2006 -- 2007), HFSP (2013 - 2017), EC Horizon 2020 ITN (2017-2020).

Related Publications
- Bornberg-Bauer E. How are model protein structures distributed in sequence space? Biophys.J. 1997 Pubmed
- E Bornberg-Bauer and HS Chan Modeling Evolutionary Landscapes: Mutational Stability, Topology and Superfunnels in Sequence Space. PNAS 1999 Online Access
- Y Cui et al. Recombinatoric exploration of novel folded structures: A heteropolymer-based model of protein evolutionary landscapes. PNAS 2002 Online Access
- R Wroe et al. A Structural Model of Latent Evolutionary Potentials Underlying Neutral Networks in Proteins. HFSP J 2007 Online Access
- D Whitehead et al. The look-ahead effect of phenotypic mutations. Biol. Dir. 2008 Online Access
- E Bornberg-Bauer et al. How do new proteins arise? Curr Opn Struct Biol. 2010 Online Access
- T Sikosek, HS Chan, E Bornberg-Bauer;. Escape from Adaptive Conflict follows from weak functional trade-offs and mutational robustness. PNAS 2012 Online Access
- Bert van Loo , Magdalena Heberlein , Philip Mair , Anastasia Zinchenko , Jan Schüürmann , Bernard D G Eenink , Josephin M Holstein, Carina Dilkaute, Joachim Jose , Florian Hollfelder, Erich Bornberg-Bauer High-Throughput, Lysis-Free Screening for Sulfatase Activity Using Escherichia coli Autodisplay in Microdroplets ACS Synthetic Biol, 8 (12), 2690-2700 Online Access
- Patrick Buchholz, Bert van Loo, Bernard Eenink, Erich Bornberg-Bauer, Jürgen Pleiss Ancestral sequences of a large promiscuous enzyme family correspond to bridges in sequence space in a network representation J. R. Soc. Interface.182021038920210389 Online Access
- Gloria Yang, Dave W Anderson, Florian Baier, Elias Dohmen, Nansook Hong, Paul D Carr, Shina Caroline Lynn Kamerlin, Colin J Jackson, Erich Bornberg-Bauer , Nobuhiko Tokuriki Higher-order Epistasis Shapes the Fitness Landscape of a Xenobiotic-Degrading Enzyme Nat Chem Biol, 15 (11), 1120-1128 Online Access
Techniques employed: computational: ancestor reconstruction, phylogenies, calculation of stability effects of mutations (e.g. FodX),
simulations of population dynamics, disorder prediction; experimental: High throughput functional molecular screening using in-cell assays and
"lab-on-a-chip" micro-droplets (Hollfelder group); experimental measuring of stability and structural dis-/order (CD); detection assays, cloning,
(over-)expression, purification, SDS page, E.coli autodisplay (Jose), differential scanning fluorimetry to study unfolding / TD stability,
ko-libraries such as ASKA (E.coli), detection assays.